Explore how Graph Neural Networks are reshaping drug discovery in 2025–2026. From molecular property prediction and drug-target interaction to de novo molecule generation and retrosynthesis — discover how GNNs are cutting costs, accelerating timelines, and finding candidates traditional methods miss entirely.
Table of Contents
Developing a new drug from scratch takes an average of 10 to 15 years and costs somewhere between one and three billion dollars.
And most of the time, it fails anyway.
More than 90% of drug candidates that enter clinical trials never make it to market. They fail because of unexpected toxicity, insufficient efficacy, poor bioavailability, or drug interactions that weren’t predicted early enough. By the time those failures happen, hundreds of millions of dollars and years of research have already been spent.
This isn’t a problem of scientific incompetence. It’s a problem of scale and complexity.
The chemical space of drug-like molecules is estimated to contain more than 10⁶⁰ possible compounds. That’s a number so large it’s essentially incomprehensible — more than the number of atoms in the observable universe. No amount of traditional laboratory screening can meaningfully explore that space.
What you need is a way to computationally predict which molecules are worth synthesizing and testing before you ever pipette a single sample.
Graph Neural Networks are becoming that tool.
In 2025–2026, GNNs are active across virtually every stage of the drug discovery pipeline — predicting molecular properties, modeling drug-target interactions, generating novel candidate molecules, planning synthesis routes, and identifying existing drugs that could be repurposed for new diseases.
The impact is real, measurable, and growing.
Why Drug Discovery Needs a Better Approach
The traditional drug discovery pipeline looks roughly like this:
Target identification — identify a biological target (usually a protein) involved in a disease. Hit discovery — screen large libraries of compounds to find ones that interact with the target. Lead optimization — refine promising hits to improve potency, selectivity, and drug-like properties. Preclinical testing — test in cell cultures and animal models for safety and efficacy. Clinical trials — test in humans across three phases. Regulatory approval — submit evidence to FDA/EMA for approval.
Each of these stages is expensive, slow, and has a high failure rate. The failures late in the pipeline — in phase II or phase III clinical trials — are the most costly.
Most of these late failures can be traced back to problems that were present in the molecule from the beginning but weren’t predicted early enough:
- toxicity that shows up in human metabolism but not in animal models
- insufficient binding affinity to the target protein
- poor solubility that prevents the drug from reaching its target
- interactions with other drugs the patient is taking
If you could predict these properties accurately before synthesis and testing, you could fail fast and cheap instead of slow and expensive. You could focus laboratory resources on the candidates most likely to succeed.
That’s exactly what GNNs are being built to do.
Why Molecules Are Graphs
The foundational insight behind GNNs for drug discovery is simple and powerful:
A molecule is a graph. Atoms are nodes. Chemical bonds are edges.
This isn’t just a convenient metaphor — it’s a mathematically natural representation. A molecule’s properties emerge from the identities of its atoms, the types of bonds between them, and the three-dimensional arrangement of those atoms and bonds. All of that information is encoded in the molecular graph.
Node features: atom type, atomic number, charge, hybridization,
aromaticity, number of hydrogens
Edge features: bond type (single/double/triple/aromatic),
bond length, ring membership, stereochemistry
Graph: the complete molecular structureBefore GNNs, computational drug discovery relied on molecular fingerprints — hand-engineered bit vectors that encode the presence or absence of specific chemical substructures. Fingerprints like ECFP (Extended Connectivity Fingerprints) are fast and widely used, but they have a fundamental limitation: they encode what a chemist thought to look for, not everything the molecule actually is.
GNNs learn representations directly from the molecular graph — without any human specification of which features matter. They discover structural patterns from data that fingerprint-based methods never capture, because nobody thought to encode them.
This is why GNNs consistently find non-obvious structure-property relationships that fingerprint-based and descriptor-based methods miss.
How GNNs Process Molecular Data
A GNN processes a molecule through repeated rounds of message passing — the same core mechanism described in the main GNN article, but applied to molecular graphs specifically.
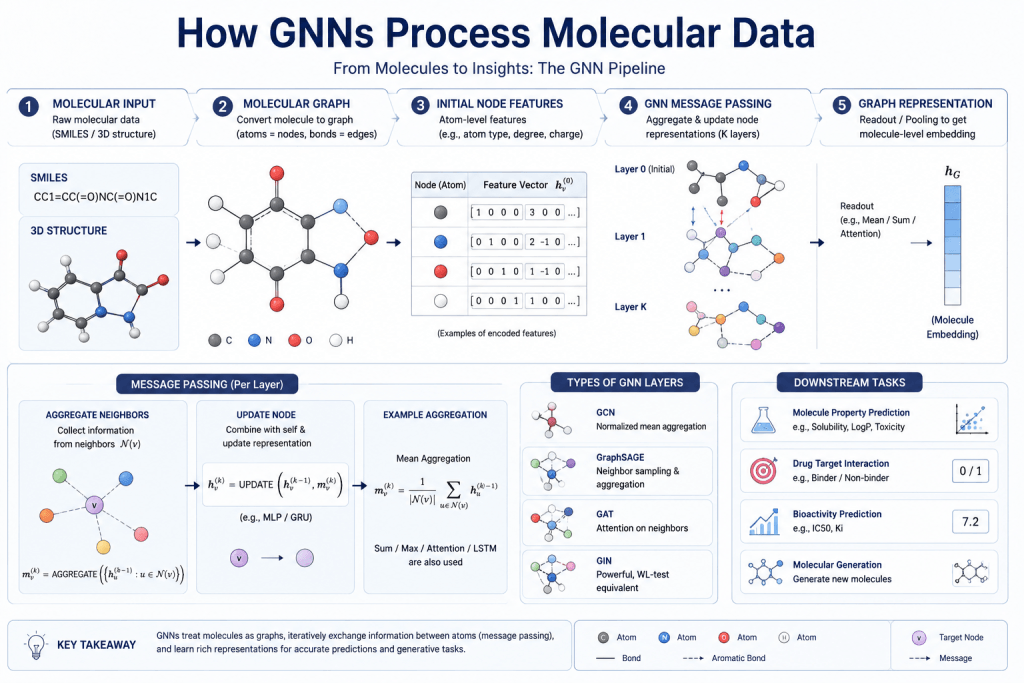
Each atom starts with a feature vector encoding its chemical properties. Through message passing, each atom iteratively updates its representation by aggregating information from its bonded neighbors:
For each atom u in the molecule:
Collect messages from all bonded atoms v
Aggregate those messages
Update atom u's representation
Repeat for k layers → k-hop chemical neighborhoodAfter several rounds of this, each atom’s representation captures not just its own chemistry but the chemical context of its local neighborhood — its bonded atoms, their bonds, the rings they’re part of, the electronic environment they create.
For molecular-level predictions (like overall solubility or toxicity), a readout function aggregates all atom representations into a single molecular fingerprint:
h_molecule = READOUT({h_u : u ∈ V})
(e.g., sum, mean, or attention-weighted pooling)That molecular embedding is then fed into a standard neural network for the final prediction task.
For atom-level predictions (like reactive sites or binding contacts), the individual atom representations are used directly.
Molecular Property Prediction
This is the most studied and most immediately useful application of GNNs in drug discovery.
Given a molecule’s structure, can we predict whether it will be soluble enough to be delivered to its target? Stable enough to survive metabolism? Potent enough to have therapeutic effect?
These questions map directly to measurable molecular properties, and GNNs have demonstrated strong predictive performance across all of them on standard benchmarks.
Key Properties GNNs Predict
Physicochemical properties
- Solubility (ESOL, FreeSolv datasets)
- Lipophilicity — how readily the drug crosses cell membranes
- Melting point and boiling point
- Molecular weight and polar surface area
Biological activity
- Whether a compound is active against a specific target (classification)
- IC₅₀ and EC₅₀ values — concentration needed for 50% inhibition or activation
- Binding affinity to specific protein targets
Safety and toxicity
- Toxicity across multiple assays (Tox21 dataset — 12 toxicity endpoints)
- hERG channel inhibition — a common cause of cardiac toxicity
- Carcinogenicity and mutagenicity (Ames test prediction)
- Blood-brain barrier penetration
How Well Do GNNs Actually Perform?
A comprehensive benchmarking study published in Artificial Intelligence in Chemistry (June 2026) systematically compared seven models — GAT, GCN, GIN, SimpleGNN, SVM, Random Forest, and XGBoost — across key physicochemical property prediction tasks.
The results are nuanced and worth understanding honestly:
On well-represented properties with large datasets, GNNs consistently outperform traditional descriptor-based methods. On smaller datasets with fewer than a few thousand compounds, traditional ML methods — particularly SVM — sometimes match or outperform GNNs because GNNs have more parameters to learn and need more data to generalize.
The practical takeaway: GNNs are strongest when you have substantial training data and when the property depends on structural patterns that are hard to hand-engineer into fingerprints. For simple physicochemical endpoints on small datasets, a well-tuned SVM is still competitive.
Benchmark datasets: ESOL, FreeSolv, Lipophilicity, MUV, HIV,
BACE, BBBP, Tox21, ToxCast, SIDER, ClinTox
Strong GNN models: Attentive FP, D-MPNN (Chemprop), GIN, GATDrug-Target Interaction Prediction
Finding that a molecule has the right properties in isolation isn’t enough. It also has to actually bind to its target — a specific protein involved in the disease you’re trying to treat.
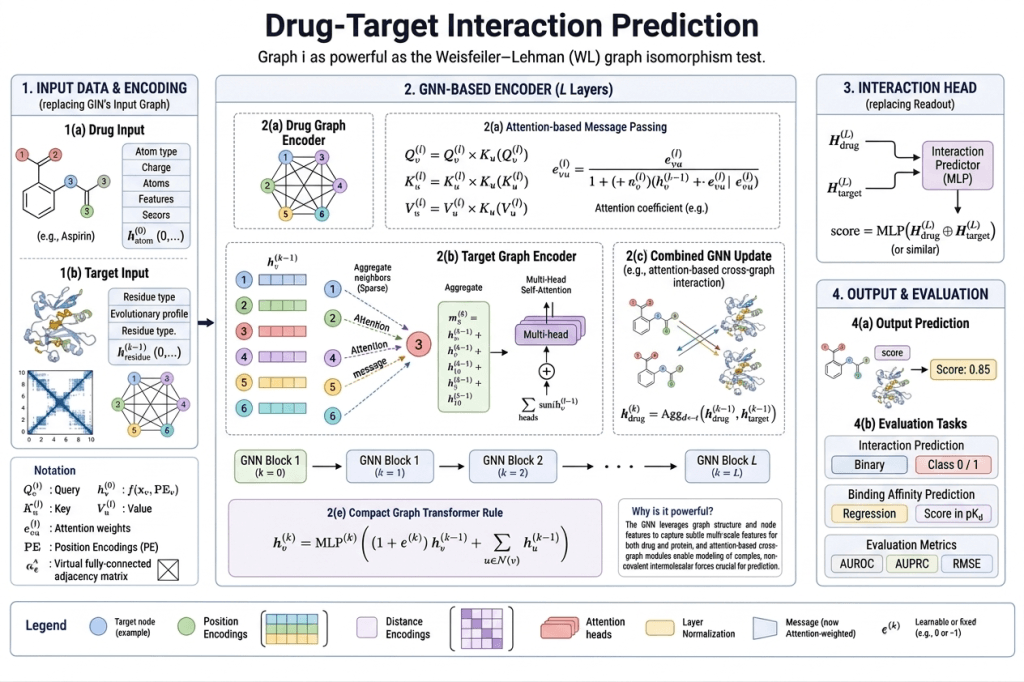
Predicting drug-target interaction (DTI) is one of the hardest problems in computational drug discovery, because it requires modeling not just the drug molecule but the target protein — and the interaction between them.
GNNs handle this naturally by representing both the drug and the target as graphs:
- The drug is a molecular graph (atoms and bonds)
- The protein is a graph of amino acids connected by peptide bonds and spatial proximity
A GNN processes both graphs, generating embeddings for each, and then a prediction head models their interaction — predicting binding affinity, docking pose, or simply whether significant binding occurs.
Recent Advances in 2025–2026
Equivariant GNNs (EGNNs) have become increasingly important for DTI prediction because they incorporate 3D atomic coordinates directly and are equivariant to rotation and translation — meaning the predicted binding affinity doesn’t change if you rotate the molecule in space, which is physically correct.
Recent models predict both protein pocket structure and drug docking within that pocket in a unified two-stage framework — using EGNN and cross-attention encoding to model the interaction between drug and target simultaneously. This approach outperforms traditional docking methods including Glide, Vina, and DiffDock on standard benchmarks.
GNN-MA (Soft Molecular Alignment with Cross-Graph Attention, 2026) introduces soft alignment between query and reference molecules for ligand-based virtual screening — identifying new active compounds against a target based on structural similarity to known actives, but with learned rather than hard-coded similarity metrics.
Key challenge: Protein structures are large and flexible — representing
them efficiently while capturing the binding site geometry
Benchmark datasets: PDBbind, Davis, KIBA, BindingDBADMET Prediction
ADMET stands for Absorption, Distribution, Metabolism, Excretion, and Toxicity — the five pharmacokinetic properties that determine whether a drug actually works in a living organism.
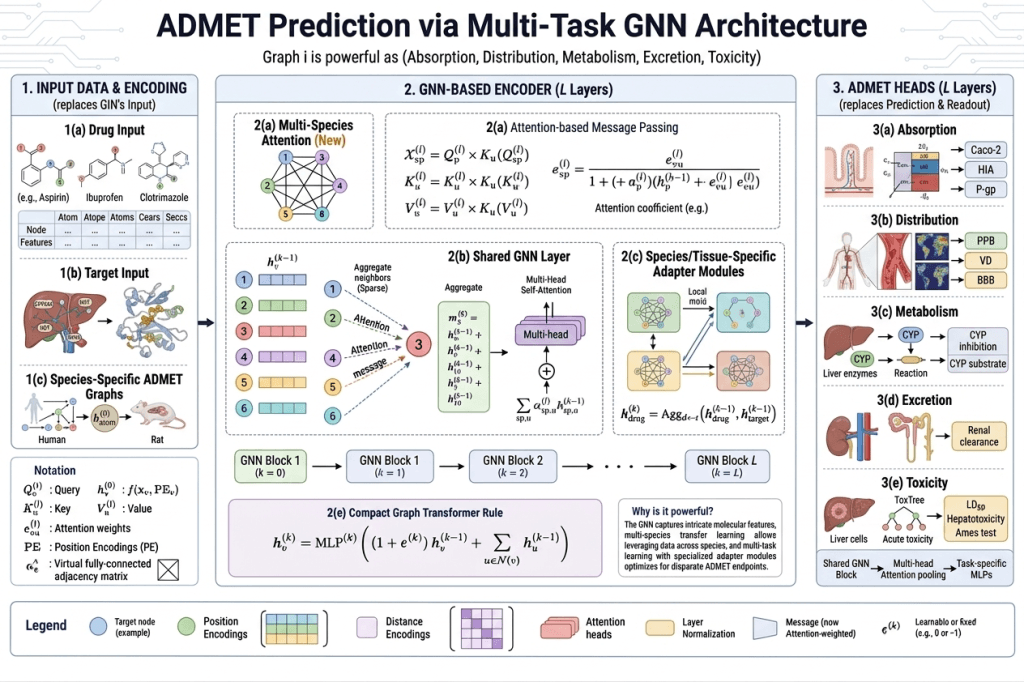
A molecule can be perfectly potent against its target in a test tube and still fail completely as a drug because:
- it doesn’t absorb well from the gut (absorption)
- it doesn’t reach the target tissue (distribution)
- it gets broken down too quickly by liver enzymes (metabolism)
- it accumulates to toxic levels instead of being cleared (excretion)
- it causes organ damage at therapeutic doses (toxicity)
ADMET failure accounts for a huge fraction of late-stage clinical trial failures — the most expensive kind. Predicting these properties computationally before synthesis is one of the highest-value applications of molecular GNNs.
GNNs learn ADMET properties from training sets of compounds with experimentally measured pharmacokinetic data. The same message-passing framework that predicts solubility can predict metabolic stability, CYP enzyme inhibition (which affects drug-drug interactions), P-glycoprotein efflux (which affects brain penetration), and organ-specific toxicity.
A Hybrid GNN-CNN framework proposed in 2025 for drug-target binding affinity prediction with quantified uncertainty is particularly relevant here — the uncertainty quantification tells medicinal chemists not just what the predicted ADMET value is, but how confident the model is. A high-uncertainty prediction flags a compound that needs experimental measurement before progressing, rather than being accepted or rejected based on an unreliable prediction.
Key ADMET endpoints predicted by GNNs:
- Oral bioavailability (F)
- Blood-brain barrier penetration (BBB)
- Half-life (t½)
- CYP450 inhibition (1A2, 2C9, 2C19, 2D6, 3A4)
- hERG cardiac toxicity
- Ames mutagenicity
- Human intestinal absorption (HIA)Drug-Drug Interaction Prediction
Most patients — especially elderly patients with multiple conditions — take more than one medication simultaneously. When two drugs interact in unexpected ways, the consequences range from reduced efficacy to life-threatening adverse events.
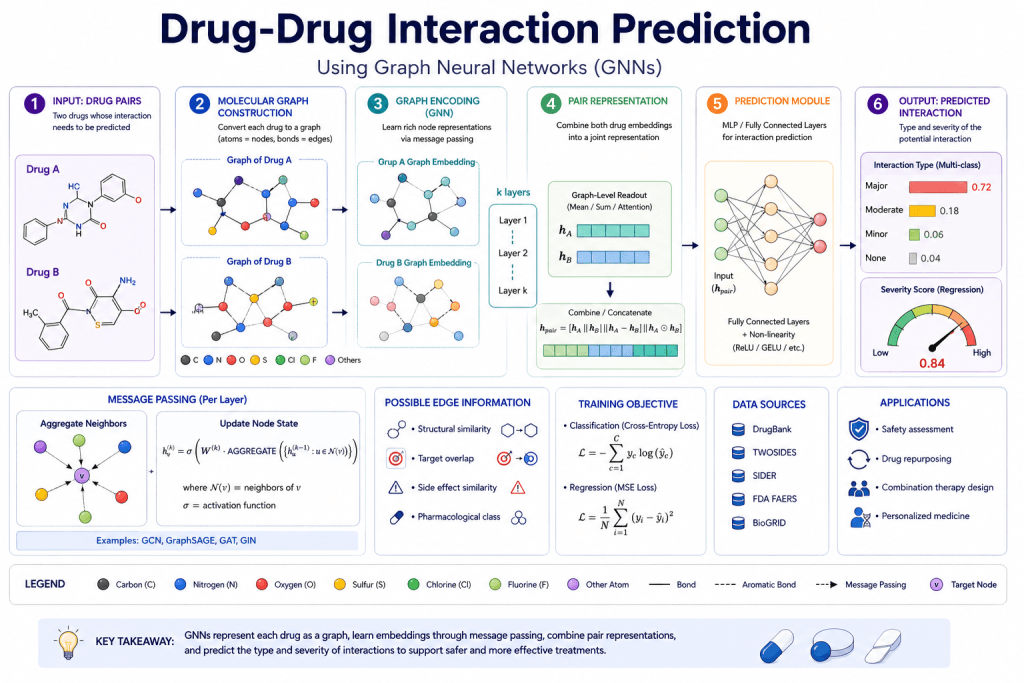
Drug-drug interaction (DDI) prediction is naturally a graph problem: drugs are nodes, known interactions are edges, and the task is to predict which unknown interactions are likely to exist.
GNNs approach this by learning molecular representations for each drug and then modeling the interaction between pairs of drugs — predicting whether an interaction occurs and what type it is (synergistic, antagonistic, or adverse).
GAINET (2025) advanced DDI prediction by combining graph attention mechanisms with interaction-specific encoding — modeling not just whether two drugs interact but the mechanism of interaction, which is critical for understanding and managing polypharmacy.
The microbiome adds another dimension: gut bacteria metabolize drugs before they reach systemic circulation, and different patients have different microbiomes. GNN models for microbiome-drug interaction prediction are an emerging area, predicting how the gut microbiome modifies a drug’s effective concentration and metabolic products.
Applications:
- Automated DDI screening for new drug candidates
- Personalized medicine — predicting interactions specific to
a patient's drug regimen
- Clinical decision support systems
Benchmark datasets: DrugBank, TWOSIDES, DeepDDI datasetDe Novo Molecule Generation
Predicting properties of existing molecules is valuable. Generating entirely new molecules with desired properties is transformative.
De novo molecule generation asks: given a desired set of properties — high binding affinity to target X, low toxicity, good oral bioavailability — can we design a molecule that has them?
The chemical space of drug-like molecules is so vast (>10⁶⁰ compounds) that random exploration is hopeless. You need a generative model that can navigate this space intelligently, proposing structurally novel molecules that are likely to have the desired properties.
GNN-based generative models approach this in several ways:
Graph-Based VAEs and GANs
Variational Autoencoders (VAEs) learn a continuous latent space of molecular graphs. Optimization in the latent space — guided by property predictions — produces new molecules with improved properties. The original MolVAE showed this was feasible; subsequent work improved the validity and novelty of generated molecules.
GAN-based approaches (MolGAN and successors) use a generator that produces molecular graphs and a discriminator that distinguishes real from fake molecules — learning to generate valid, drug-like structures.
Fragment-Enhanced Generation
A 2025 approach combines fragment-enhanced GNNs with Conditional Variational Autoencoders (CVAE) for target-specific molecular generation. Rather than generating molecules atom by atom, it works at the level of chemically meaningful fragments (motifs) — pieces of molecular structure that are likely to be synthetically accessible and drug-like.
The two-stage framework first pre-trains on unlabeled molecular data through self-supervised learning, then incorporates protein pocket information as a condition for generating molecules specifically designed to fit a target’s binding site. This significantly outperforms atom-level generation approaches on property prediction benchmarks.
Diffusion-Based Molecule Generation
The success of diffusion models in image generation has inspired molecular diffusion models — treating molecule generation as a denoising process in 3D space. These models generate 3D molecular structures directly, with atom positions and bond types emerging from the denoising process.
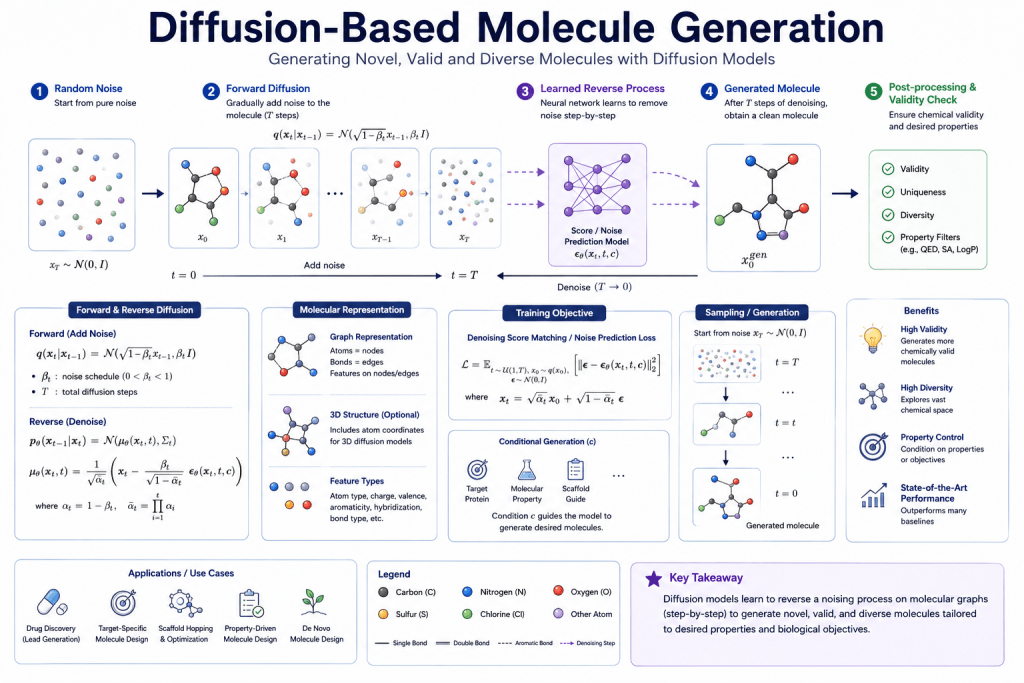
In 2025–2026, diffusion-based molecular generation (DiffSBDD, TargetDiff, and successors) is producing highly promising results for structure-based drug design — generating molecules specifically shaped to occupy a protein binding pocket.
Evaluation metrics for molecular generation:
- Validity: % of generated molecules that are chemically valid
- Uniqueness: % of generated molecules that are non-duplicate
- Novelty: % not present in training data
- Drug-likeness: QED score, SA score (synthetic accessibility)
- Property optimization: binding affinity, ADMET complianceRetrosynthesis Planning
Designing a molecule with good predicted properties is only half the problem.
You also have to be able to synthesize it.
Retrosynthesis is the process of working backwards from a target molecule to identify a sequence of chemical reactions that could produce it from commercially available starting materials. This is one of the most cognitively demanding tasks in medicinal chemistry — experienced chemists spend years developing the intuition to plan synthetic routes efficiently.
GNNs are learning to do this computationally.
Template-based retrosynthesis GNNs learn to identify which reaction templates from known chemical databases apply to a given target molecule — predicting the most likely retrosynthetic steps based on learned patterns from millions of known reactions.
Template-free retrosynthesis More powerful and more flexible — GNNs predict which bonds in the target molecule to break and how to transform the fragments, without being constrained to a fixed template library. This can suggest novel synthetic routes that human chemists wouldn’t have considered.
Graph-based reaction prediction Systems like GraphRXN represent chemical reactions as graph transformations — predicting the products of forward reactions and the precursors of retrosynthetic steps in a unified framework.
3D geometric GNNs incorporating atomic coordinates predict regioselectivity — which specific site on a molecule a reaction will occur at — enabling chemists to design site-selective transformations with improved efficiency. This is particularly valuable for late-stage functionalization of complex drug scaffolds.
Benchmark datasets: USPTO-50K, USPTO-FULL, Reaxys
Key metrics: Top-1 accuracy, Top-5 accuracy (are correct precursors
in the top 5 predictions?)Drug Repositioning
Developing a completely new drug from scratch is the slow, expensive path.
Drug repositioning — finding new therapeutic uses for drugs that are already approved for other indications — is dramatically faster and cheaper because the safety profile of the repositioned drug is already established. You skip the early safety phases entirely.
GNNs approach drug repositioning through knowledge graphs — large graphs that encode known relationships between drugs, diseases, genes, proteins, side effects, and biological pathways.
The task is link prediction: given a drug node and a disease node in the knowledge graph, predict whether an undiscovered therapeutic relationship exists between them — based on the pattern of existing connections in the graph.
This is how GNNs identified existing drugs as potential candidates for COVID-19 treatment in 2020 — by finding patterns of connectivity in drug-disease-protein graphs that suggested unexplored therapeutic relationships.
In 2025–2026, deep learning in stroke therapeutics is an active drug repurposing area — GNNs identifying existing approved drugs whose mechanism of action may be protective against stroke pathology based on biological pathway graphs.
GNNs for natural product discovery are another emerging area — predicting which natural compounds with known structures might have activity against cancer metastasis and chemoresistance, based on structural similarity to known actives and biological pathway connectivity.
Key knowledge graph sources: DrugBank, STRING, DisGeNET,
OMIM, KEGG, UniProt
Task: Link prediction between drug and disease nodesKey GNN Architectures Used in Drug Discovery
Not all GNN architectures are equally suited to molecular tasks. Here’s how the major ones perform in drug discovery contexts:
| Architecture | Key Strength | Drug Discovery Application |
|---|---|---|
| GCN | Fast, simple baseline | Initial property screening |
| GAT | Attention on important bonds | Binding affinity, DTI |
| GIN | Maximum expressiveness | Graph-level classification |
| MPNN | Explicit edge features | Reaction prediction, ADMET |
| Attentive FP | Atom-level attention | Multi-task property prediction |
| D-MPNN (Chemprop) | Directed message passing | General molecular property |
| EGNN | 3D equivariance | Structure-based drug design |
| Graph Transformer | Long-range interactions | Macromolecular modeling |
Attentive FP — which combines graph attention with a novel readout mechanism — consistently performs among the strongest models on the MoleculeNet benchmark across multiple property prediction tasks.
D-MPNN (Chemprop) uses directed message passing — treating bonds as directed edges — which improves performance by distinguishing the direction of information flow through the molecular graph. It’s one of the most widely used GNN frameworks in pharmaceutical research.
EGNNs are increasingly important for 3D molecular modeling because they respect the physical symmetries of molecular systems — predictions don’t change when you rotate or translate the molecule in space.
Benchmark Datasets
| Dataset | Task | Size | Description |
|---|---|---|---|
| ESOL | Solubility prediction | 1,128 | Aqueous solubility |
| FreeSolv | Hydration free energy | 643 | Experimental and calculated |
| Lipophilicity | Lipophilicity | 4,200 | Experimental octanol/water |
| HIV | Antiviral activity | 41,127 | Active vs inactive classification |
| BACE | Beta-secretase inhibition | 1,513 | Alzheimer’s target |
| BBBP | Blood-brain barrier | 2,039 | CNS drug penetration |
| Tox21 | Multi-task toxicity | 7,831 | 12 toxicity endpoints |
| ToxCast | Toxicity assays | 8,575 | 617 assay endpoints |
| MUV | Virtual screening | 93,087 | Challenging actives/decoys |
| QM9 | Quantum properties | 134,000 | DFT-computed properties |
| PDBbind | Binding affinity | 19,443 | Protein-ligand complexes |
| ZINC | Drug-like molecules | 250,000+ | Purchasable compounds |
MoleculeNet (Wu et al., 2018) standardized these datasets into a unified benchmarking framework — making fair comparisons between GNN architectures possible. Most published GNN papers report results on MoleculeNet datasets.
OGB-molhiv and OGB-molpcba from the Open Graph Benchmark provide larger, more challenging molecular property prediction tasks that better differentiate between architectures.
GNN vs Traditional ML for Molecular Property Prediction
This is a question that deserves an honest answer rather than a GNN advocacy piece.
The 2026 benchmarking study in Artificial Intelligence in Chemistry compared GNNs against classical methods systematically:
Where GNNs win:
- Large datasets (thousands+ of compounds)
- Properties that depend on complex structural patterns not captured by standard fingerprints
- Multi-task learning — GNNs share representations across related tasks, improving performance when individual task data is limited
- 3D structure-dependent properties where geometric information matters
Where traditional ML is competitive:
- Small datasets (< 1,000 compounds) — GNNs overfit without sufficient data
- Simple physicochemical properties well-captured by existing descriptors
- Computational speed — SVMs and random forests are faster at inference
- Interpretability — SVM models with fingerprint features are easier to interpret
The practical recommendation: For a new molecular property prediction task, run both a GNN (D-MPNN/Chemprop is a good default) and a gradient boosting model on ECFP fingerprints. Use the GNN when data is abundant and the property is structurally complex. Use the traditional model when data is limited or interpretability is critical.
The best industrial workflows use both — with GNNs handling the deep structural pattern recognition and traditional methods providing interpretable baselines.
Research Trends in 2025–2026
3D Geometric and Equivariant GNNs
Standard GNNs process molecular graphs in 2D — atom connectivity without 3D coordinates. But molecular properties depend critically on 3D structure — the shape of a molecule determines how it fits into a protein binding pocket, how it interacts with metabolic enzymes, and how it crosses cell membranes.
Equivariant GNNs (SE(3)-GNNs, EGNNs, PaiNN, NequIP) incorporate 3D atomic coordinates directly and are designed to be equivariant to rotation and translation — physically meaningful symmetries that 2D GNNs ignore. These models are producing the strongest results on structure-based drug design tasks in 2025–2026.
Pre-training and Transfer Learning
Training molecular GNNs from scratch requires large labeled datasets — which are expensive and time-consuming to generate experimentally.
Pre-training GNNs on large unlabeled molecular datasets (millions of molecules from ZINC or PubChem) through self-supervised tasks — predicting masked atom types, predicting molecular properties from partial observations — then fine-tuning on small labeled datasets for specific properties, is dramatically improving performance in low-data regimes.
This mirrors what BERT and GPT did for NLP — pre-training on unlabeled data creates general representations that transfer efficiently to downstream tasks with limited labels.
The fragment-enhanced GNN approach (2025) uses exactly this strategy — self-supervised pre-training on unlabeled molecular data, followed by fine-tuning for target-specific generation.
GNN and LLM Integration for Drug Discovery
Large language models trained on chemical literature and molecular sequences (like ChemBERTa, MolGPT, and their successors) are being combined with GNNs in hybrid architectures.
The combination is powerful:
- LLMs understand chemical language — IUPAC names, reaction descriptions, biological context from literature
- GNNs understand molecular structure — topology, geometry, and learned structure-property relationships
Combining both produces models that can reason about molecules in the context of their biological and chemical literature — understanding not just the structure but the experimental history of similar compounds.
Uncertainty Quantification
For drug discovery applications, knowing how confident a model is matters enormously.
A GNN that predicts a compound will have good ADMET properties should also report its confidence in that prediction. Low confidence should trigger experimental measurement rather than accepting the prediction. High-uncertainty predictions in silico are not a reliable basis for expensive synthesis decisions.
Bayesian GNNs, ensemble methods, and conformal prediction approaches are increasingly used in 2025–2026 pharmaceutical applications — providing calibrated uncertainty estimates alongside property predictions.
Multi-Task Learning
Drug discovery requires predicting many properties simultaneously — and they’re not independent. A modification that improves potency often worsens solubility. A change that reduces toxicity may affect metabolic stability.
Multi-task GNNs learn all of these properties simultaneously from shared molecular representations — allowing related tasks to share information and improving performance on individual tasks, especially when some have limited training data.
Tox21 (12 toxicity endpoints) and ToxCast (617 assay endpoints) are canonical multi-task drug discovery benchmarks where this approach shows clear benefits.
Challenges and Open Problems
Data scarcity for specialized targets For many novel disease targets, experimental data is limited. GNNs need data to learn, and the most interesting drug discovery problems often have the least available training data. Pre-training and transfer learning help but don’t fully solve the problem.
Synthesizability A GNN can generate molecules with excellent predicted properties that are practically impossible to synthesize in a laboratory. Incorporating synthetic accessibility constraints into generative models — ensuring generated molecules can actually be made — remains an active challenge.
Interpretability When a GNN predicts that a molecule will be toxic, identifying which structural features caused that prediction is important for medicinal chemists who need to modify the molecule to fix the problem. GNN explanations (GNNExplainer, attention weights) provide partial answers but aren’t always chemically meaningful.
Distribution shift A GNN trained on existing drug-like molecules may perform poorly when applied to structurally novel chemical series outside its training distribution. As generative models push into new chemical space, this becomes increasingly important.
3D structure availability Equivariant GNNs are powerful but require 3D molecular conformations as input. Generating and validating 3D structures adds computational overhead and introduces errors when predicted conformations differ from true bound conformations.
Experimental validation gap Computational predictions need laboratory validation. The feedback loop between GNN predictions and experimental measurements is long and expensive. Shortening this loop — through active learning, automated synthesis, and high-throughput screening — is essential for realizing the full potential of GNN-guided drug discovery.
Final Thoughts
The promise of AI-accelerated drug discovery has been talked about for a long time.
What’s different in 2025–2026 is that it’s actually happening — not as a futuristic possibility but as a working part of pharmaceutical research pipelines at major companies.
GNNs have earned their place in this pipeline because they’re genuinely better than the alternatives at specific, important tasks. They find structure-property relationships that fingerprint-based methods miss. They model drug-target interactions in ways that docking scores can’t fully capture. They generate novel molecules that occupy previously unexplored chemical space. They plan synthesis routes that experienced chemists validate as feasible.
None of this replaces experimental chemistry. The laboratory is still where molecules prove or disprove their value. But GNNs are changing what gets to the laboratory — which candidates get synthesized and tested, and in what order.
That shift — from random screening to intelligent, model-guided selection — is where the real value is.
If GNNs can improve the hit rate in early-stage discovery by even a modest amount, the downstream effect on clinical trial success rates and drug development timelines is enormous. The molecules selected by better models fail earlier, fail cheaper, and sometimes don’t fail at all.
That’s the actual transformation happening in drug discovery right now. Not GNNs replacing chemists — GNNs helping chemists spend their time on experiments worth doing.
FAQ
How are GNNs used in drug discovery?
GNNs are used across multiple stages of drug discovery: predicting molecular properties (solubility, toxicity, binding affinity), modeling drug-target interactions, predicting ADMET pharmacokinetic properties, generating novel drug candidate molecules, planning retrosynthesis routes, and identifying existing drugs for repositioning to new indications.
Why are molecules represented as graphs?
Molecules are naturally graph structures — atoms are nodes and chemical bonds are edges. This representation captures the topology and geometry of molecular structure directly, without requiring hand-engineered features. GNNs learn from this structure end-to-end, discovering structure-property relationships that fingerprint-based methods miss.
What is ADMET prediction in drug discovery?
ADMET stands for Absorption, Distribution, Metabolism, Excretion, and Toxicity — the five pharmacokinetic properties that determine whether a drug works safely in a living organism. GNNs predict these properties computationally from molecular structure before synthesis, helping prioritize candidates that are likely to succeed in laboratory and clinical testing.
What is de novo molecule generation?
De novo molecule generation is the computational design of entirely new molecules with desired properties. GNN-based generative models — VAEs, GANs, and diffusion models operating on molecular graphs — navigate the vast chemical space to propose novel drug candidates with high binding affinity, good ADMET properties, and synthetic accessibility. This is particularly powerful for targets where no good starting molecules are known.
What is retrosynthesis in drug discovery?
Retrosynthesis is working backwards from a target molecule to identify which chemical reactions could produce it from commercially available starting materials. GNNs learn to predict retrosynthetic steps from millions of known chemical reactions — suggesting synthesis routes that medicinal chemists can evaluate and execute in the laboratory.
Are GNNs better than traditional ML for molecular property prediction?
It depends on the task and data availability. GNNs outperform traditional ML (SVM, Random Forest) on large datasets and structurally complex properties. On small datasets (under 1,000 compounds) or simple physicochemical endpoints, traditional methods are often competitive or superior. The best approach is to benchmark both for each specific task.
What is drug repositioning and how do GNNs help?
Drug repositioning identifies new therapeutic uses for existing approved drugs — a faster and cheaper path than developing new drugs from scratch, because safety is already established. GNNs model drug-disease relationships as link prediction in large biological knowledge graphs, identifying unexpected therapeutic connections between existing drugs and diseases based on patterns of biological pathway connectivity.
What benchmark datasets are used for GNN drug discovery research?
The most widely used benchmarks are MoleculeNet (which standardizes datasets including Tox21, HIV, BACE, BBBP, ESOL, FreeSolv, and Lipophilicity), QM9 for quantum chemical properties, PDBbind for protein-ligand binding affinity, and the Open Graph Benchmark (OGB) molecular datasets for larger-scale evaluation.